MindSponge分子動力學模擬——使用迭代器進行系統演化(2023.09)
技術背景
在前面幾篇部落格中,我們已經介紹過使用MindSponge去定義一個系統以及使用MindSponge計算一個分子系統的單點能。這篇文章我們將介紹一下在MindSponge中定義迭代器Updater,並使用Sponge對系統進行演化,最後使用CallBack對輸出結果進行追蹤和儲存。
分子動力學迭代器與深度學習優化器
首先我們回顧一下這張MindSponge的軟體架構圖:
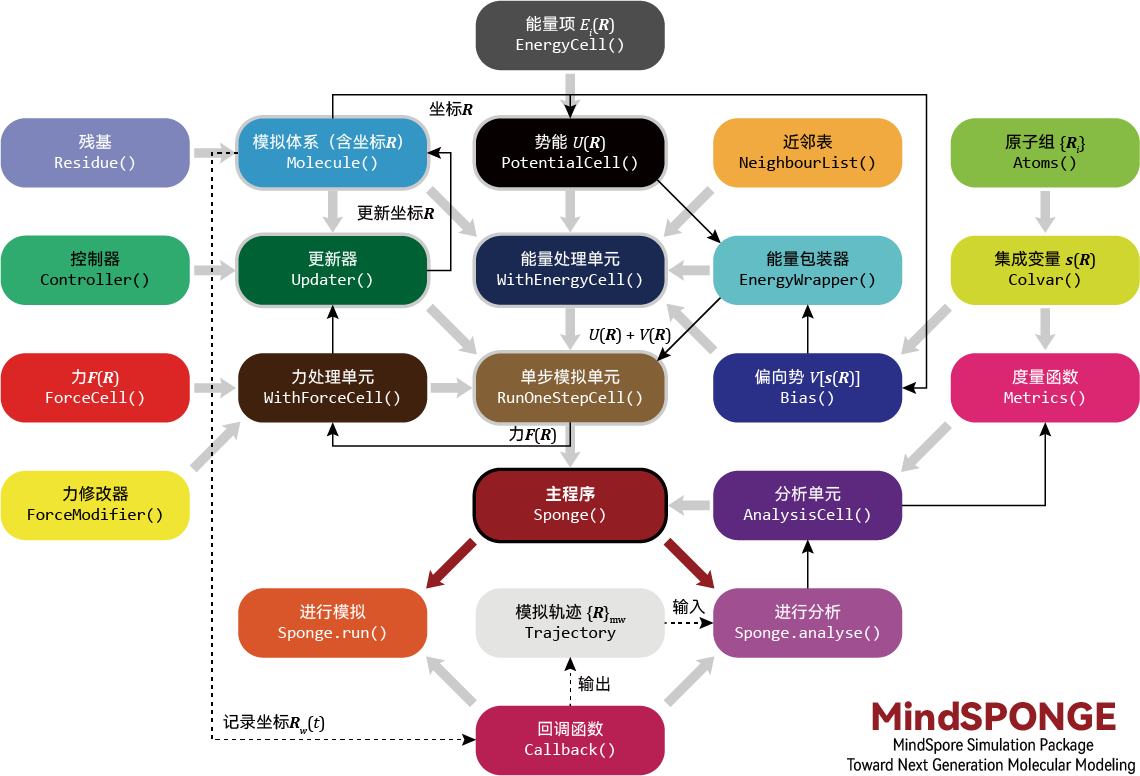
在這裡Updater從WithForceCell接收一個力(這個力可以是直接從外界輸入的,也可以是利用MindSpore的自動微分功能,對WithEnergyCell進行自動微分得到的力),並將其作用在Molecule系統上,得到一個Molecule新的狀態。這個流程可以對照深度學習裡面的優化器,比如梯度下降法,我們也是從損失函數中計算一個梯度(依賴於自動微分或者差分),然後將這個梯度根據不同的優化演演算法計算一個迭代值,最後作用在網路引數上,得到一組新的網路引數。所以,分子動力學中的迭代器跟深度學習中的優化器,本質上可以是相同的,換句話說,我們可以直接使用深度學習中的一些優化演演算法(如Adam等)來作為分子動力學模擬中的迭代器。比如說,我們可以參考如下方案做一個能量極小化。
能量極小化
其實做能量極小化的思路是簡單的,我們假設單點能\(E(R)\)(維度為[B,1])是一個關於原子座標\(R\)(維度為[B,A,D])的一個函數。那麼所謂的能量極小化,就是找到這樣的一個最低能量值:
假定我們就使用MindSpore中內建的Adam演演算法,那麼相應的程式碼實現可以參考如下案例。首先我們有一個用於模擬的pdb案例檔案:
REMARK Generated By Xponge (Molecule)
ATOM 1 N ALA 1 -0.095 -11.436 -0.780
ATOM 2 CA ALA 1 -0.171 -10.015 -0.507
ATOM 3 CB ALA 1 1.201 -9.359 -0.628
ATOM 4 C ALA 1 -1.107 -9.319 -1.485
ATOM 5 O ALA 1 -1.682 -9.960 -2.362
ATOM 6 N ARG 2 -1.303 -8.037 -1.397
ATOM 7 CA ARG 2 -2.194 -7.375 -2.328
ATOM 8 CB ARG 2 -3.606 -7.943 -2.235
ATOM 9 CG ARG 2 -4.510 -7.221 -3.228
ATOM 10 CD ARG 2 -5.923 -7.789 -3.136
ATOM 11 NE ARG 2 -6.831 -7.111 -4.087
ATOM 12 CZ ARG 2 -8.119 -7.421 -4.205
ATOM 13 NH1 ARG 2 -8.686 -8.371 -3.468
ATOM 14 NH2 ARG 2 -8.844 -6.747 -5.093
ATOM 15 C ARG 2 -2.273 -5.882 -2.042
ATOM 16 O ARG 2 -1.630 -5.388 -1.119
ATOM 17 N ALA 3 -3.027 -5.119 -2.777
ATOM 18 CA ALA 3 -3.103 -3.697 -2.505
ATOM 19 CB ALA 3 -1.731 -3.041 -2.625
ATOM 20 C ALA 3 -4.039 -3.001 -3.483
ATOM 21 O ALA 3 -4.614 -3.643 -4.359
ATOM 22 N ALA 4 -4.235 -1.719 -3.394
ATOM 23 CA ALA 4 -5.126 -1.057 -4.325
ATOM 24 CB ALA 4 -6.538 -1.625 -4.233
ATOM 25 C ALA 4 -5.205 0.436 -4.039
ATOM 26 O ALA 4 -4.561 0.930 -3.116
ATOM 27 OXT ALA 4 -5.915 1.166 -4.728
TER
然後可以用MindSponge實現一個簡單的用Adam進行能量極小化的程式碼:
from mindspore import nn, context
from sponge import ForceField, Sponge, set_global_units, Protein
from sponge.callback import RunInfo, WriteH5MD
# 設定MindSpore的執行環境
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
# 設定全域性單位
set_global_units('A', 'kcal/mol')
# 定義一個基於case1.pdb的分子系統
system = Protein('case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
# 定義一個amber.ff99sb的力場
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定義一個學習率為1e-03的Adam優化器
min_opt = nn.Adam(system.trainable_params(), 1e-03)
# 定義一個用於執行分子模擬的Sponge範例
md = Sponge(system, potential=energy, optimizer=min_opt)
# RunInfo這個回撥函數可以在螢幕上根據指定頻次輸出能量引數
run_info = RunInfo(200)
# WriteH5MD回撥函數,可以將軌跡、能量、力和速度等引數保留到一個hdf5檔案中,檔案字尾為h5md
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False)
# 開始執行分子動力學模擬,執行2000次迭代
md.run(2000, callbacks=[run_info, cb_h5md])
上述程式碼的執行結果如下:
[MindSPONGE] Adding 45 hydrogen atoms for the protein molecule in 0.003 seconds.
[MindSPONGE] Started simulation at 2023-09-04 15:14:29
[MindSPONGE] Step: 0, E_pot: 1200.4639
[MindSPONGE] Step: 200, E_pot: 7.763489
[MindSPONGE] Step: 400, E_pot: -70.34643
[MindSPONGE] Step: 600, E_pot: -96.88522
[MindSPONGE] Step: 800, E_pot: -109.98717
[MindSPONGE] Step: 1000, E_pot: -117.33747
[MindSPONGE] Step: 1200, E_pot: -121.95378
[MindSPONGE] Step: 1400, E_pot: -125.20764
[MindSPONGE] Step: 1600, E_pot: -127.72044
[MindSPONGE] Step: 1800, E_pot: -129.79828
[MindSPONGE] Finished simulation at 2023-09-04 15:15:16
[MindSPONGE] Simulation time: 46.79 seconds.
--------------------------------------------------------------------------------
此時我們可以看到整個分子能量是一直在下降的,同時在該路徑下生成了一個test.h5md的軌跡檔案。開啟這個軌跡檔案的方式有兩種,一種是使用silx view test.h5md(可以使用python3 -m pip install silx來進行安裝)來檢視檔案的具體內容,比如可以看到這樣的一個結果:
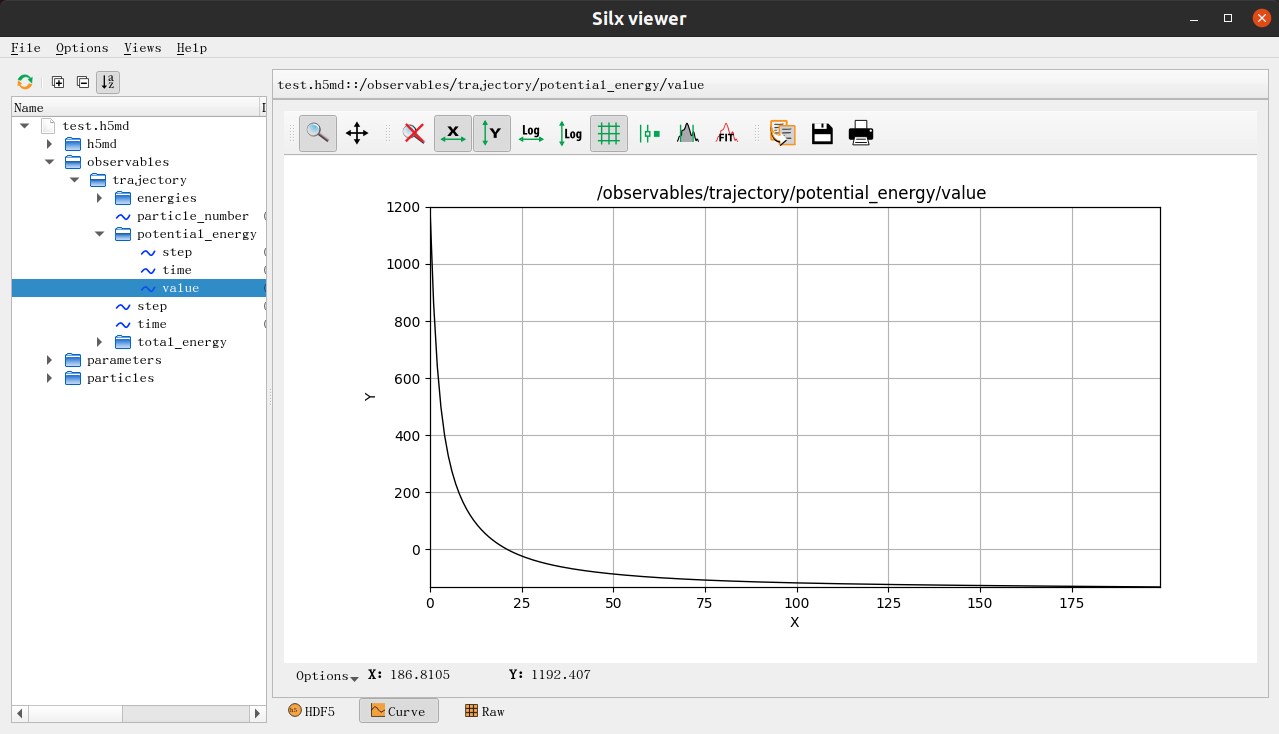
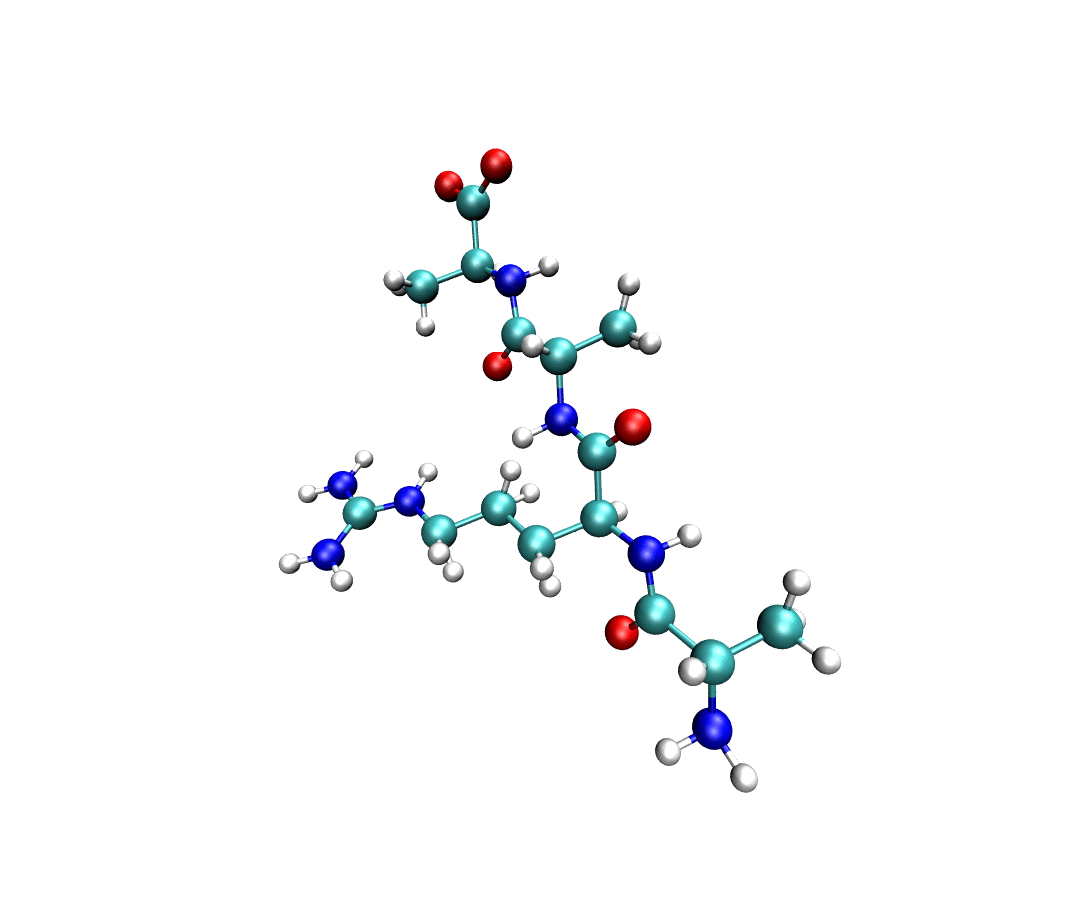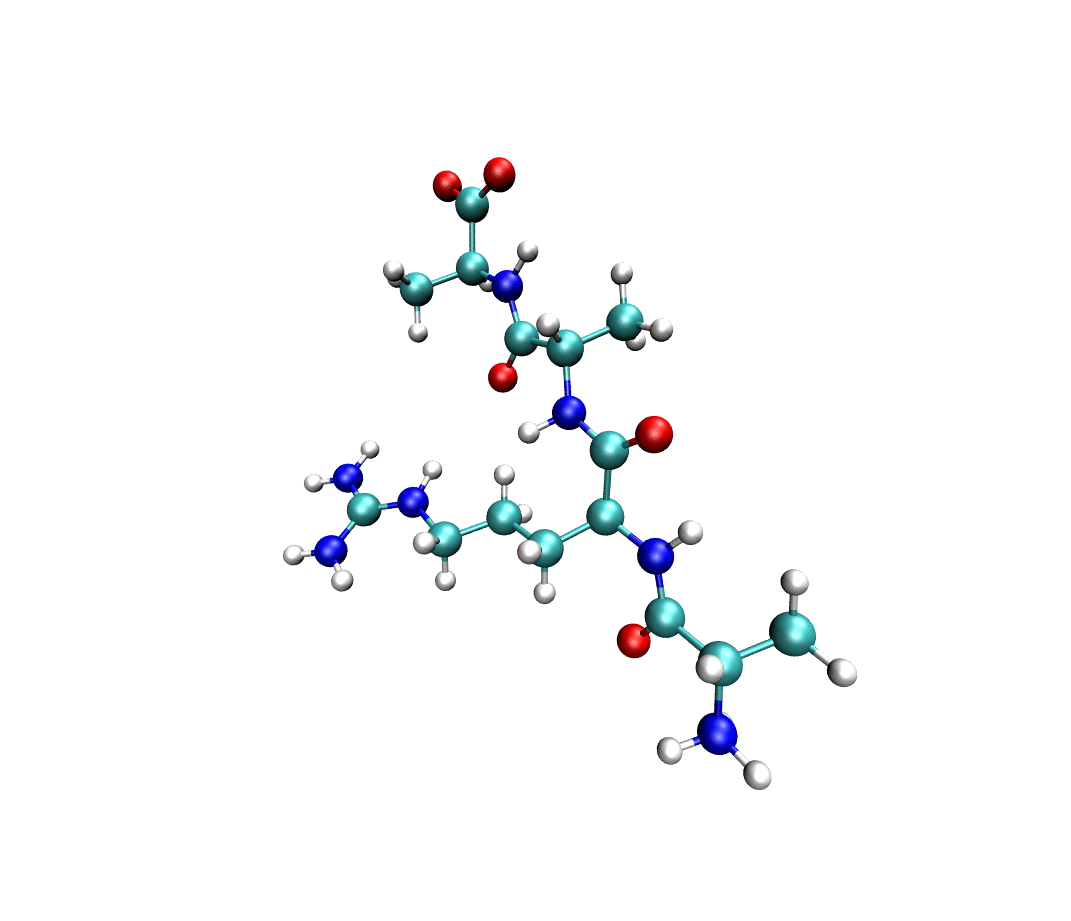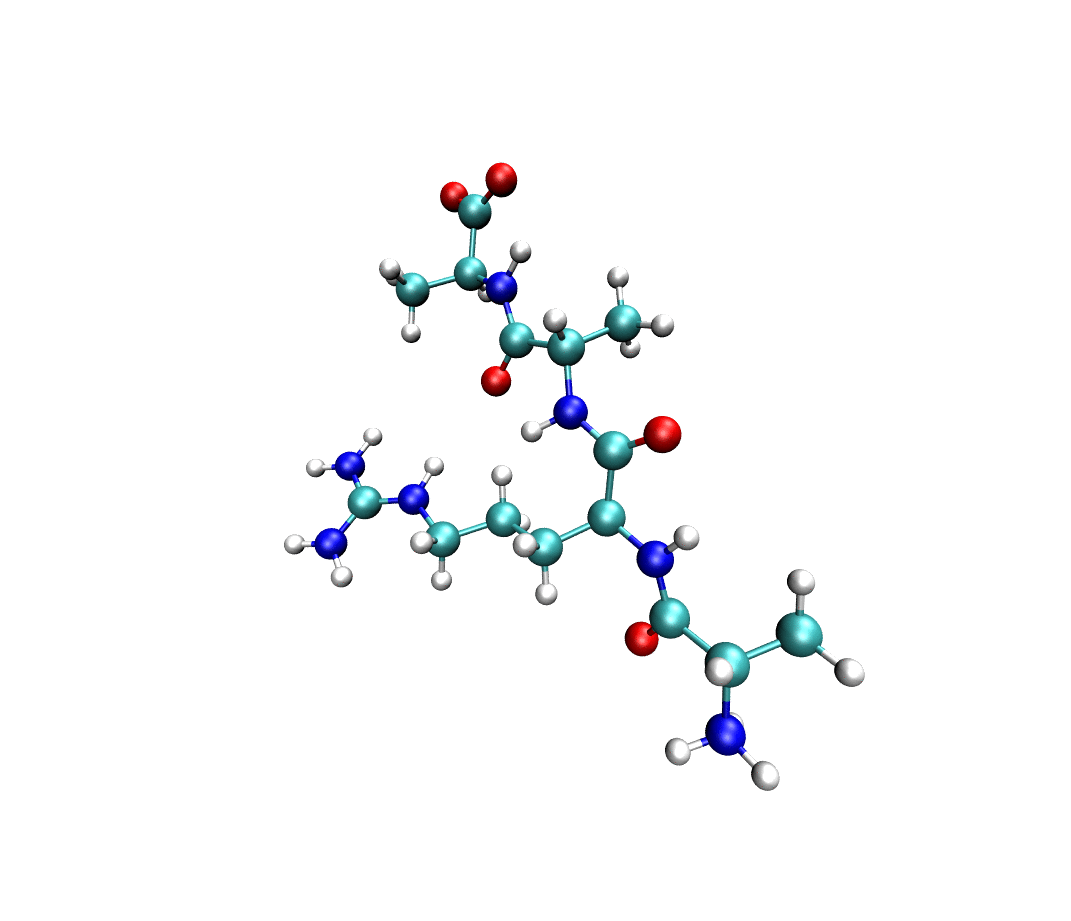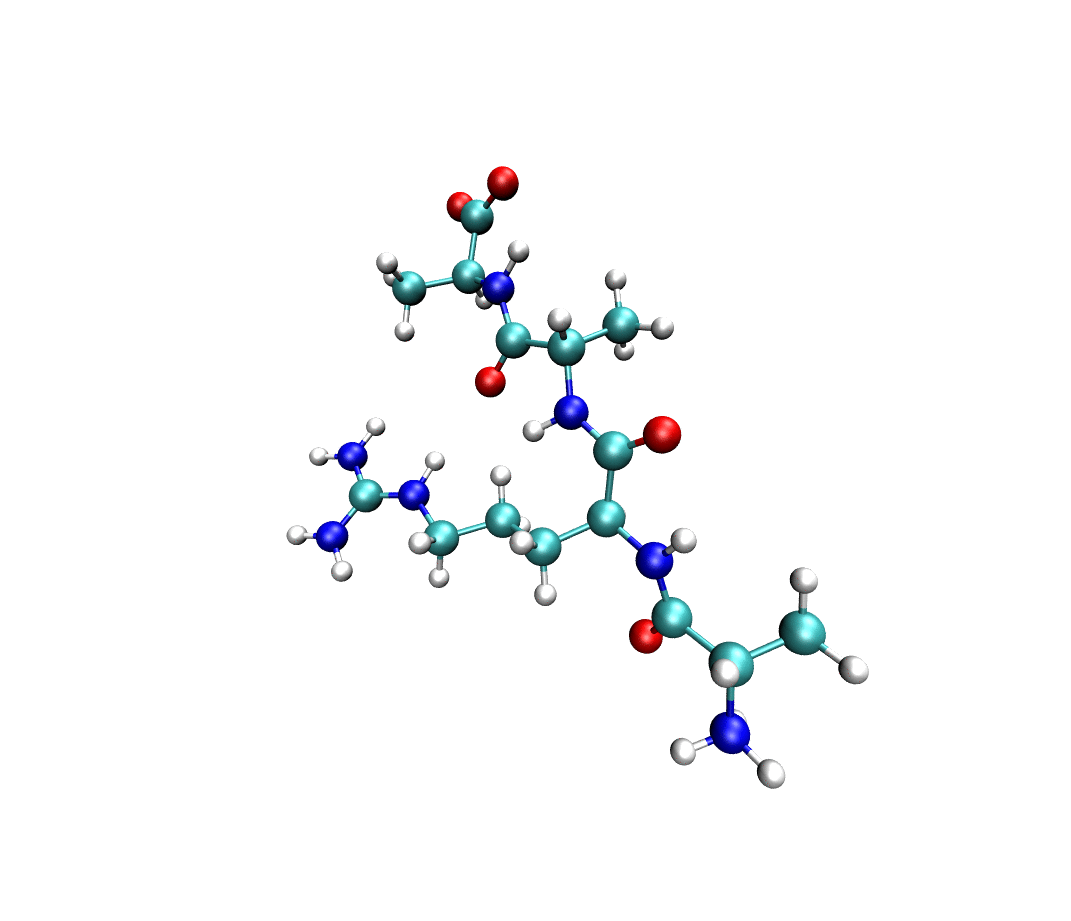
既可以使用曲線圖的方式來進行瀏覽,也可以使用表格的方式來進行瀏覽。而如果參考這個README.md檔案的指示安裝一個VMD外掛的話,就可以在本地直接用VMD來視覺化分子模擬的軌跡,輸出為gif動態圖如下所示:
分子動力學模擬
在上一個章節中,我們演示了一下使用MindSpore中自帶的優化器做了一個能量極小化,得到了一個穩定的構象。需要說明的是,在上一步的過程中,如果我們想保留最後一幀的結果,既可以使用VMD匯出,也可以在WriteH5MD中進行相應的引數設定,比如在上面的案例中將對應程式碼修改為:
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False, save_last_pdb='case1_min.pdb')
就可以在執行結束之後生成一個case1_min.pdb檔案。因為在上一步的執行過程中我們已經對氫原子進行了重構,因此這裡如果我們重新執行一個動力學模擬的任務的話,可以不需要重構氫原子,對應的程式碼應調整為:
system = Protein('case1_min.pdb', template=['protein0.yaml'], rebuild_hydrogen=False)
那麼最終整體的程式碼如下所示:
from mindspore import context
from sponge import ForceField, Sponge, set_global_units, Protein, UpdaterMD, WithEnergyCell, RunOneStepCell
from sponge.function import VelocityGenerator
from sponge.callback import RunInfo, WriteH5MD
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
set_global_units('A', 'kcal/mol')
# 這裡設定rebuild_hydrogen為False,意為不對氫原子進行重構
system = Protein('case1_min.pdb', template=['protein0.yaml'], rebuild_hydrogen=False)
energy = ForceField(system, parameters=['AMBER.FF99SB'])
# 定義一個速度生成器,使得生成的隨機速度可以讓系統處在300K的溫度下
vgen = VelocityGenerator(300)
# 根據速度生成器生成相應的原子速度
velocity = vgen(system.shape, system.atom_mass)
# UpdaterMD迭代器,這裡給定了temperature和thermostat,是一個NVT過程,積分器使用的是velocity_verlet演演算法
opt = UpdaterMD(system=system,
time_step=1e-3,
velocity=velocity,
integrator='velocity_verlet',
temperature=300,
thermostat='langevin')
# 定義Sponge可以有很多種方法,這裡採用的是RunOneStep來定義
sim = WithEnergyCell(system, energy)
one_step = RunOneStepCell(energy=sim, optimizer=opt)
md = Sponge(one_step)
run_info = RunInfo(200)
cb_h5md = WriteH5MD(system, 'test.h5md', save_freq=10, write_image=False)
md.run(2000, callbacks=[run_info, cb_h5md])
除了前面提到的兩處程式碼修改之外,其他的調整見程式碼中的註釋。上述程式碼的執行結果為:
[MindSPONGE] Started simulation at 2023-09-04 17:06:26
[MindSPONGE] Step: 0, E_pot: -131.60876, E_kin: 52.25413, E_tot: -79.35463, Temperature: 313.0393
[MindSPONGE] Step: 200, E_pot: -112.29764, E_kin: 39.081543, E_tot: -73.216095, Temperature: 234.12614
[MindSPONGE] Step: 400, E_pot: -125.55388, E_kin: 66.579636, E_tot: -58.974243, Temperature: 398.85922
[MindSPONGE] Step: 600, E_pot: -127.8087, E_kin: 59.09694, E_tot: -68.71176, Temperature: 354.03256
[MindSPONGE] Step: 800, E_pot: -150.88245, E_kin: 69.84598, E_tot: -81.03647, Temperature: 418.42694
[MindSPONGE] Step: 1000, E_pot: -163.9544, E_kin: 62.79237, E_tot: -101.16203, Temperature: 376.1708
[MindSPONGE] Step: 1200, E_pot: -159.15376, E_kin: 55.752754, E_tot: -103.40101, Temperature: 333.99854
[MindSPONGE] Step: 1400, E_pot: -159.43027, E_kin: 57.967392, E_tot: -101.462875, Temperature: 347.26575
[MindSPONGE] Step: 1600, E_pot: -163.72491, E_kin: 57.850487, E_tot: -105.87443, Temperature: 346.56543
[MindSPONGE] Step: 1800, E_pot: -168.06078, E_kin: 54.730705, E_tot: -113.33007, Temperature: 327.87573
[MindSPONGE] Finished simulation at 2023-09-04 17:07:13
[MindSPONGE] Simulation time: 47.41 seconds.
--------------------------------------------------------------------------------
因為上面這個案例我們執行的是一個NVT恆溫過程,因此我們可以用silx view看到,在結果中所儲存的溫度最終會逐漸趨近於300K附近:
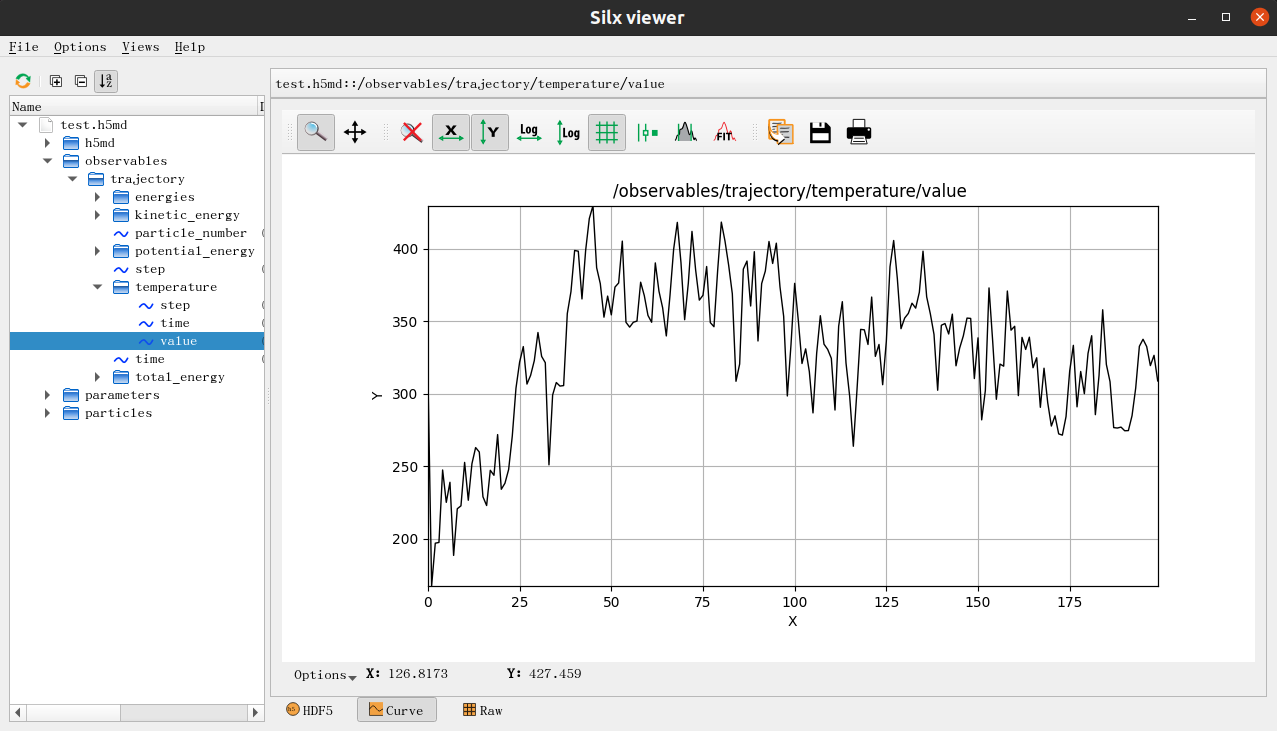
同樣的,我們可以用VMD的外掛來視覺化這個分子運動的軌跡:
迭代器切換
之所以採用Python這一程式語言來實現,很大程度上就考慮到了各種方法實現的便捷性。比如上述章節中定義好一個Sponge範例之後,我們需要切換其中的優化器——這其實是一個比較常用的方法,例如我們執行完了一個能量極小化的過程,我們甚至都不需要退出程式,直接用演化好的system可以繼續執行NVT過程,然後執行NPT過程。而這一系列的操作只需要用到一個函數:change_optimizer。
from mindspore import nn, context
from sponge import ForceField, Sponge, set_global_units, Protein, UpdaterMD
from sponge.function import VelocityGenerator
from sponge.callback import RunInfo, WriteH5MD
context.set_context(mode=context.GRAPH_MODE, device_target='GPU', device_id=1)
set_global_units('A', 'kcal/mol')
system = Protein('case1.pdb', template=['protein0.yaml'], rebuild_hydrogen=True)
energy = ForceField(system, parameters=['AMBER.FF99SB'])
min_opt = nn.Adam(system.trainable_params(), 1e-03)
md = Sponge(system, potential=energy, optimizer=min_opt)
run_info = RunInfo(200)
cb_h5md = WriteH5MD(system, 'test_min.h5md', save_freq=10, write_image=False)
md.run(2000, callbacks=[run_info, cb_h5md])
# 定義一個新的迭代器,並用change_optimizer完成切換
vgen = VelocityGenerator(300)
velocity = vgen(system.shape, system.atom_mass)
opt = UpdaterMD(system=system,
time_step=1e-3,
velocity=velocity,
integrator='velocity_verlet',
temperature=300,
thermostat='langevin')
md.change_optimizer(opt)
nvt_h5md = WriteH5MD(system, 'test_nvt.h5md', save_freq=10, write_image=False)
md.run(2000, callbacks=[run_info, nvt_h5md])
上述程式碼的執行結果如下:
[MindSPONGE] Adding 45 hydrogen atoms for the protein molecule in 0.003 seconds.
[MindSPONGE] Started simulation at 2023-09-04 17:29:29
[MindSPONGE] Step: 0, E_pot: 1200.4639
[MindSPONGE] Step: 200, E_pot: 7.7634506
[MindSPONGE] Step: 400, E_pot: -70.34645
[MindSPONGE] Step: 600, E_pot: -96.885216
[MindSPONGE] Step: 800, E_pot: -109.987114
[MindSPONGE] Step: 1000, E_pot: -117.33747
[MindSPONGE] Step: 1200, E_pot: -121.95381
[MindSPONGE] Step: 1400, E_pot: -125.20767
[MindSPONGE] Step: 1600, E_pot: -127.72041
[MindSPONGE] Step: 1800, E_pot: -129.79825
[MindSPONGE] Finished simulation at 2023-09-04 17:30:13
[MindSPONGE] Simulation time: 43.96 seconds.
--------------------------------------------------------------------------------
[MindSPONGE] Started simulation at 2023-09-04 17:30:14
[MindSPONGE] Step: 0, E_pot: -131.61736, E_kin: 58.91965, E_tot: -72.69771, Temperature: 352.9705
[MindSPONGE] Step: 200, E_pot: -114.52379, E_kin: 45.360672, E_tot: -69.16312, Temperature: 271.74258
[MindSPONGE] Step: 400, E_pot: -120.40167, E_kin: 48.916946, E_tot: -71.484726, Temperature: 293.04718
[MindSPONGE] Step: 600, E_pot: -127.64978, E_kin: 60.41433, E_tot: -67.23545, Temperature: 361.92465
[MindSPONGE] Step: 800, E_pot: -152.59546, E_kin: 72.86487, E_tot: -79.73059, Temperature: 436.5122
[MindSPONGE] Step: 1000, E_pot: -152.563, E_kin: 71.374565, E_tot: -81.18844, Temperature: 427.58426
[MindSPONGE] Step: 1200, E_pot: -160.1132, E_kin: 68.66432, E_tot: -91.44888, Temperature: 411.34796
[MindSPONGE] Step: 1400, E_pot: -152.07262, E_kin: 58.346176, E_tot: -93.72644, Temperature: 349.53497
[MindSPONGE] Step: 1600, E_pot: -152.71495, E_kin: 50.630737, E_tot: -102.08421, Temperature: 303.314
[MindSPONGE] Step: 1800, E_pot: -148.00838, E_kin: 52.72198, E_tot: -95.28639, Temperature: 315.84204
[MindSPONGE] Finished simulation at 2023-09-04 17:31:00
[MindSPONGE] Simulation time: 46.71 seconds.
--------------------------------------------------------------------------------
總結概要
在經過前面幾篇部落格的介紹之後,我們可以定義一些目標的分子體系,並且計算其單點能。而分子模擬的精髓就在於快速的迭代和演化,也就是本文所要介紹的迭代器相關的內容。在具備了分子系統、單點能和迭代器這三者之後,就可以正式開始進行分子動力學模擬。常見的模擬過程有:能量極小化、NVT恆溫恆容過程、NPT恆溫恆壓過程以及NVE微正則系綜,本文所涉及的主要是能量極小化以及NVT恆溫恆容過程,更多的模擬方法有待大家一起研究探討。
版權宣告
本文首發連結為:https://www.cnblogs.com/dechinphy/p/updater-md.html
作者ID:DechinPhy
更多原著文章:https://www.cnblogs.com/dechinphy/
請博主喝咖啡:https://www.cnblogs.com/dechinphy/gallery/image/379634.html