高通量測序的資料處理與分析指北(二)-宏基因組篇
宏基因組篇
前言
之前的一篇文章已經從生物實驗的角度講述了高通量測序的原理,這篇文章旨在介紹宏基因組二代測序資料的處理方式及其原理。在正文開始之前,我們先來認識一下什麼是宏基因組。以我的理解,宏基因組就是某環境中所有生物的基因組的合集,這個環境可以是下水道,河流等自然環境,也可以是人體內腸道,口腔等體環境。而宏基因組中的生物往往指的是微生物,如真菌,細菌,病毒,古細菌。
我們這裡主要以腸道微生物為例,也就是人體內腸道的宏基因組。腸道菌群的測序樣本往往是糞便樣本,現在主流的測序方式有兩種:一種是16sRNA測序,一種是WGS(Whole Genome Sequencing) 全基因組測序。WGS測序資料量更大,所包含的資訊更多,能註釋出物種-樣本的丰度矩陣,也能註釋出基因-樣本的丰度矩陣。而16sRNA測序測的是細菌核糖體RNA中的小亞基,這個小亞基的沉降係數是 16s,故被稱為 16s RNA,這個16s RNA有一段非常保守的序列和一段變異序列,可以根據16s RNA 的變異度來進行物種分類,所以16s RNA資料往往只能註釋出物種-樣本的丰度矩陣。
原理介紹
之前文章中也提到了,由於測序技術的限制,目前二代測序只能測較短的鹼基片段,所以需要對基因進行碎片化,我們要思考的問題就是這些碎片化的基因如何重新拼回到完整的基因組或者這些碎片化的基因如何確定其屬於什麼物種從而得到物種的丰度矩陣。
目前對宏基因組原始資料如何註釋到物種的方法有兩類主流方法,一類是基於bin進行物種註釋的方法,一類是不基於bin進行物種註釋的方法
基於bin的物種註釋
基於bin的物種註釋的代表軟體有 metawrap,metabat2等。
在宏基因組的原始資料也就是fastq資料中,含有大量的read序列,首先是將read按照序列拼接成contigs,如圖所示,上面的的read按照序列重合程度拼接成下面的contigs。

然後把相類似的contigs歸為一個bin,而具體如何歸bin的方法各種軟體所用的原理都有些區別,這裡介紹兩種方法,也是這個視訊中提到的兩種分類的方法,第一種是依據四鹼基頻率來進行區分,所謂四鹼基頻率就是ATGC四個鹼基為一組,共256種鹼基組合,同一種物種的這256鹼基組合的頻率是相似的,並且物種親緣關係越遠則四鹼基頻率差距越大,故這一個256維的向量可以進行PCA降維,然後用聚類方法將類似的contigs聚到一起作為一個bin。
第二種方法是基於測序深度的,他的基本原理是由於不同的物種基因組大小不同,而同一種物種的基因組大小是類似的,因此可以根據contigs的深度來判斷其是否為同一個物種,物種的基因組越大,在隨機打碎DNA時產生的碎片越多,read數越高,最後通過read拼接而成的contigs的深度越大。
總而言之,bin就是一堆親緣關係較近的contigs的合集,也可以視為一個物種基因組的草圖。
得到高質量bin後就是對他進行基於資料庫的註釋,將能註釋出來的bin註釋出來。而bin的丰度,也就是物種的丰度的計算方式就是bin上每個鹼基的深度除以bin序列長度。這個計算方式不太確定,推測的,暫時沒找到資料
不基於bin的物種註釋
基因bin的物種註釋更加準確,但是耗時更長,這裡再介紹一類直接從read比對資料得到物種丰度的宏基因組資料處理的方式,代表的軟體有 kraken,metaphlan等。
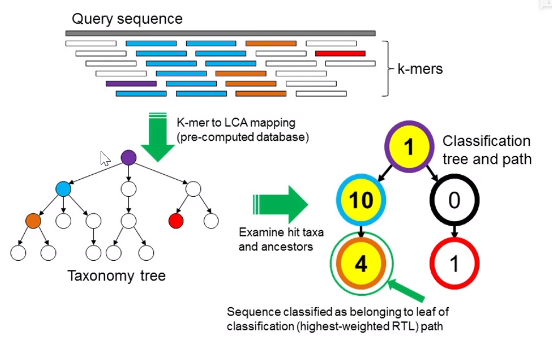
這裡主要以kraken的原理為例,它實際上就是將read 分成了多個 k-mers。 這個k-mers的意思就是是k bp長度的子序列,只不過這個子序列覆蓋了read所有鹼基,如150bp的read 能拆分出 150-31+1 個 31-mer, k-mers中的k長度是自定義的,預設是31,然後將這些 k-mers去跟資料庫比對,k-mers對上最多的分支就作為這個read的物種分類,如上圖,這個序列就是被認為是來自與4號物種的序列。同樣的,將每個物種比對上的read數量除以其基因組長度就得到了其丰度。